
近日,我所化学反应动力学全国重点实验室反应动力学理论与计算研究组(1113组)傅碧娜研究员、张东辉院士团队在多原子碰撞反应机制研究方面取得新进展,发现在Cl+C2H2→C2H+HCl反应中,占主导地位的机制为“漫游(roaming)”机制,而非传统过渡态的直接抽取机制。
长期以来,化学界普遍认为化学反应遵循传统过渡态理论,沿着最小能量路径进行。然而,近年来非传统反应途径如漫游机制逐渐受到关注。本工作聚焦的 Cl+C2H2反应,在大气化学中具有重要意义,是乙炔在海洋和极地对流层的去除途径,也是平流层下部卤素原子的重要消耗途径。
研究团队利用自主开发的基本不变量-神经网络(FI-NN)方法(J. Chem. Phys.,2020;Nat. Commun.,2022;Natl. Sci. Rev.,2023;J. Chem. Theory Comput.,2025),构建了Cl+C2H2反应的高精度全维势能面,并开展精确的动力学模拟。研究发现,一旦超过能量阈值,Cl+C2H2→C2H+HCl反应主要通过氯漫游(Cl-roaming)和氢漫游(H-roaming)两种机制进行(贡献接近100%),传统过渡态的直接抽取机制的贡献微乎其微。在 Cl-roaming 过程中,Cl 原子与乙炔形成短暂的C2H2Cl 加合物,随后Cl原子部分解离并在乙炔分子周围漫游,最终提取氢原子形成HCl;H-roaming机制则是氢原子先部分脱离,在C2HCl中间体周围迁移,进而提取氯原子生成产物。通过对反应时间、产物角分布和能量分布的深入分析,研究团队进一步揭示了该反应中漫游机制占主导地位。
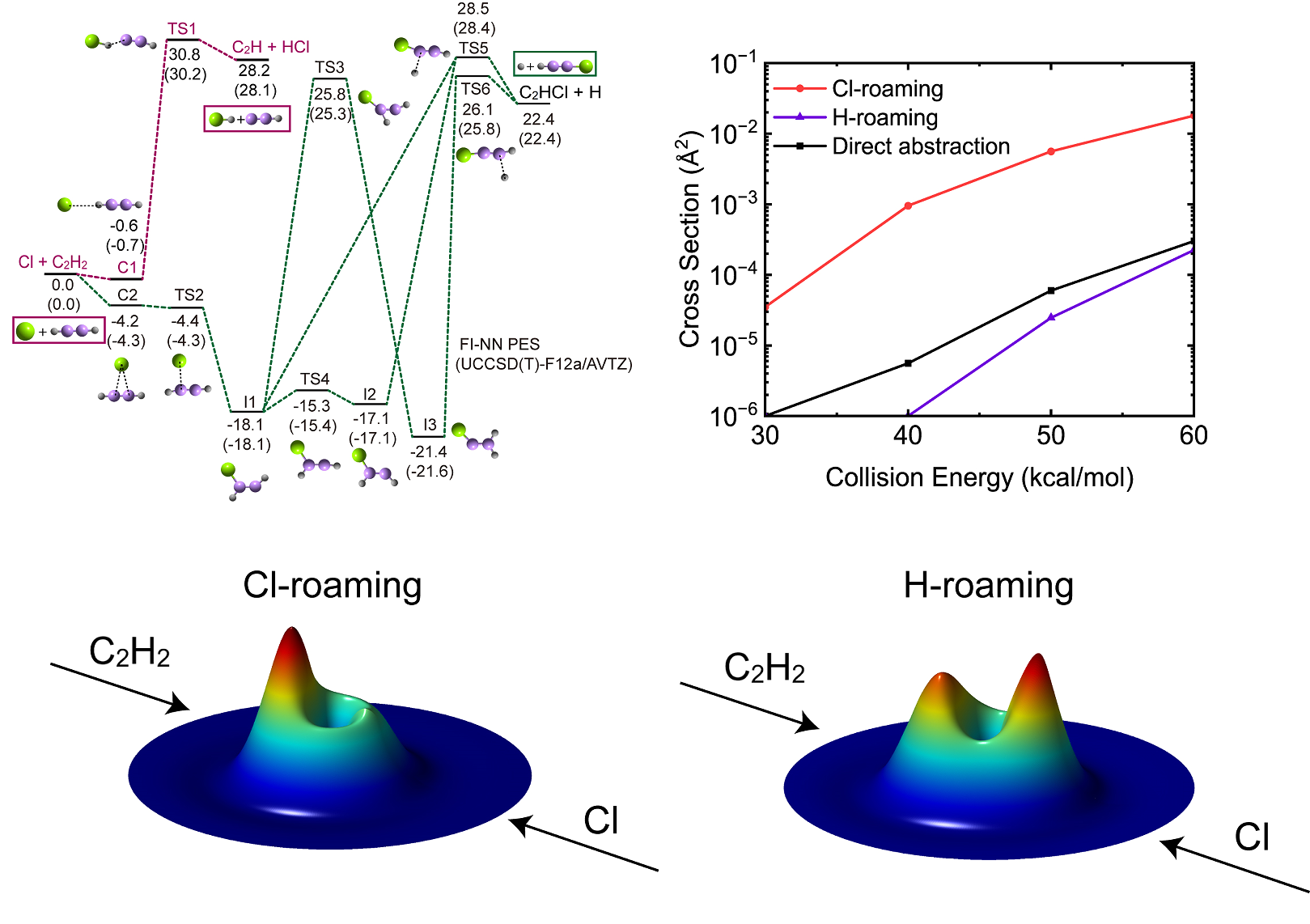
本工作不仅挑战了传统双分子反应理论,强调了在反应动力学研究中,考虑非传统途径(如漫游机制)的重要性,还为准确预测反应速率常数、深入理解反应机制提供了新视角。
傅碧娜和张东辉团队前期在漫游机制的研究中取得系列进展,提出了包括高激发态分子的漫游体系(Science,2024)、碰撞诱导漫游体系(Chem. Sci.,2020),双漫游反应体系(J. Phys. Chem. Lett.,2021)、OH自由基漫游体系(J. Phys. Chem. Lett.,2025;J. Phys. Chem. A,2023)、多电子态非绝热漫游体系(J. Am. Chem. Soc.,2011;Proc. Natl. Acad. Sci.,2012;J. Chem. Theory Comput.,2013)等,证实了漫游机制在化学反应中的普适性,为预测和理解复杂化学反应提供了理论支撑。
相关研究成果以“Exclusive roaming mechanism for the Cl + C2H2→C2H + HCl bimolecular reaction”为题,于近日发表在《自然-通讯》(Nature Communications)上。该工作的共同第一作者是我所1113组联合培养博士研究生白玉瑶和付艳林副研究员。该工作得到了国家自然科学基金、中国科学院B类先导专项“基于极紫外光源的化学反应过渡态精准探测”、科技部科技创新2030-重大项目等项目的支持。(文/图 白玉瑶、付艳林)


