

近日,我所太阳能研究部(DNL16)李灿院士、范峰滔研究员等联合厦门大学李剑锋教授团队,在水氧化反应的电化学成像研究中取得新进展。研究团队利用自主研发的高空间分辨原位电化学成像技术,并结合原位谱学表征,发现水氧化反应并非由固定不变的静态活性位点简单主导,而是由表面电荷积累驱动活性中心发生动态结构适应,进而调控不同晶面的反应路径和动力学行为。该工作为突破太阳能光(电)催化反应的“瓶颈”提供了全新的机理认识。
太阳能光(电)催化水分解能够提供清洁可再生的太阳燃料,是科学研究中的“圣杯”式反应,受到全世界关注。其中,水氧化反应涉及多个电子和质子的协同转移,是制约整体效率提升的关键步骤。然而,该反应发生在复杂的固—液界面:光生电荷在催化剂表面积累、转移,并进一步驱动表面化学键的形成与断裂。长期以来,在真实反应环境中直接观察表面电荷、活性位点和反应中间物种之间的动态耦合关系存在困难,这也成为理解水氧化反应机制的主要障碍。
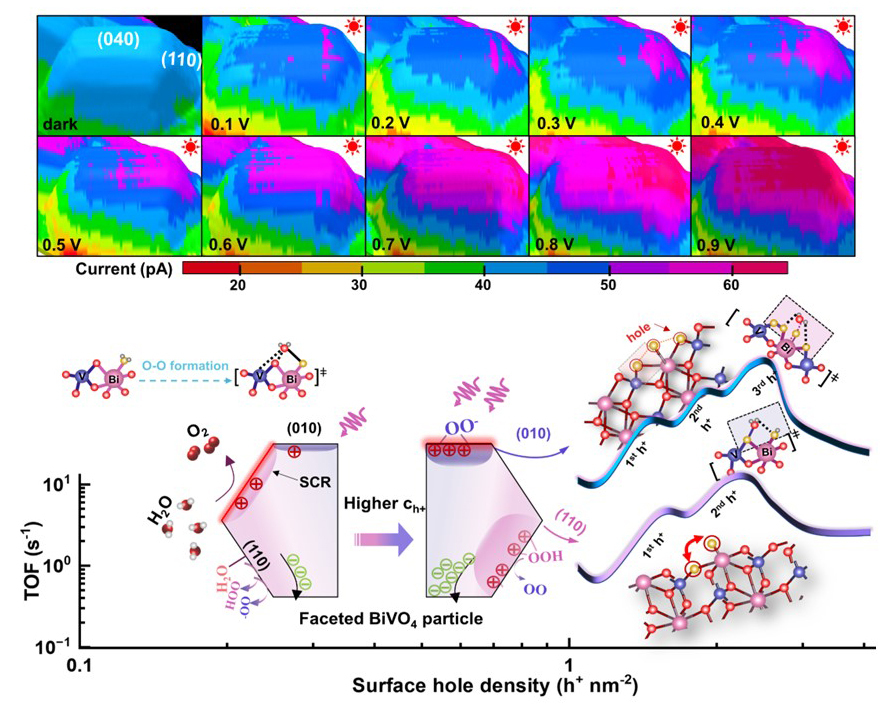
针对这一问题,研究团队发展了纳米空间分辨的原位电化学成像方法,在液相反应条件下对异质纳米催化剂表面的局域反应活性和动力学过程进行了可视化追踪。通过外球电子转移反应成像,研究人员实现了液相中催化剂表面电荷浓度的定量化“滴定”,为解析水氧化反应动力学提供了关键基础。
研究发现,水氧化反应并不是由固定不变的静态活性位点简单主导,而是由表面空穴浓度驱动的动态过程。当表面空穴密度较低时,催化剂的(110)和(010)晶面均处于单空穴转移限制的动力学区间,并稳定生成过氧羟基和过氧中间体;此时,(110)晶面表现出略高的本征活性。当表面空穴密度达到临界阈值后,反应机制发生显著转变:(010)晶面通过过氧中间体在Bi-O-V核心结构中实现动态空穴积累,催化活性迅速跃升,并表现出高阶动力学特征,其活性超过(110)晶面成为主导活性面。同时,(110)晶面则倾向于积累双氧化等价物,促进分子内O-O偶联过程。
该研究表明,高效水氧化反应依赖于多空穴协同积累机制。空穴不仅是反应所需的“电荷载体”,还参与调控催化剂表面结构,使活性中心在反应过程中发生动态适应。这一认识推动了水氧化催化机制从传统的“静态活性位点模型”转向“电荷介导的动态活性系统模型”,为下一代人工光合作用催化剂设计提供了新思路:不仅需要优化材料的静态结构,还应精准调控光生电荷与催化剂结构之间的动态耦合。
李灿、范峰滔团队围绕光催化体系中光生电荷分离与转移动力学这一核心科学问题,开展了长期而系统的全时空尺度表征研究:从晶面电荷分离(Nat. Commun.,2013)和异相结电荷分离(Acc. Chem. Res.,2025)等基础科学问题出发,自主研发了可直接表征催化剂表面光生电荷分布的表面光电压成像技术,先后实现了对晶面电荷分离(Angew. Chem. Int. Ed.,2015)、相结(J. Phys. Chem. Lett.,2017)、扩散过程(Nat. Energy,2018),以及铁电退极化场(Nat. Commun.,2025)等多尺度不同电荷分离驱动力的可视化表征,阐明了内建电场、浓度梯度和极化场等因素在空间上驱动电荷迁移的作用机制;进一步发展了时空分辨表面光电压技术,实现了光生电荷分离动力学在全时域范围内的追踪(Nature,2022;Nat. Protoc.,2024),发现了单颗粒光催化剂中多重复杂电荷转移机制和准弹道输运机制;在此基础上,揭示了光生电荷转移与催化反应位点精准耦合,从而实现高量子效率光催化完全分解水的微观机制(Nat. Commun.,2026)。近期,该团队进一步实现了固—液界面表面光生电荷的直接成像,揭示了溶剂环境对电荷分离的重要影响(J. Am. Chem. Soc.,2025,J. Am. Chem. Soc.,2025)。本次工作将成像对象从“电荷分离与转移”推进到“真实液相界面反应过程”,是团队构建人工光合作用全时空成像研究体系中的重要环节突破。
相关研究成果以“Spatial imaging of water oxidation on single-particle catalysts”为题,发表在《自然—纳米技术》(Nature Nanotechnology)上。该工作的第一作者为DNL1600组群博士毕业生聂伟。该工作得到国家自然科学基金、国家自然科学基金委“人工光合成”基础科学中心项目、国家重点研发计划、中国科学院稳定支持基础研究领域青年团队、中央高校基本科研业务费专项资金、新基石科学基金会科学探索奖等项目的资助。(文/图 聂伟、范峰滔)


