
近日,我所分子反应动力学国家重点实验室在分子表面散射动力学理论研究上获得新进展。由该实验室傅碧娜副研究员、张东辉研究员等撰写的论文“First-principles quantum dynamical theory for the dissociative chemisorption of H2O on rigid Cu(111)”发表在近期的《自然·通讯》杂志上(Nature Communications, 2016, 7:11953, doi: 10.1038/ncomms11953),该研究工作首次实现了多原子分子在金属表面反应的全维量子动力学计算。
分子在金属表面解离吸附的动力学研究在多相催化等工业过程中占有重要的地位。在过去的20多年里,科学家们为发展可靠的理论来精确描述分子在固体表面的解离吸附动力学付出了巨大的努力。由于反应中可能存在的量子效应,如量子隧穿、零点能、反应共振等,量子动力学研究是最为可靠的。但是由于高维量子动力学研究的困难,以往精确的量子动力学理论只局限于研究双原子分子在固体表面解离吸附这类包含六个自由度的问题。水在过渡态金属表面的解离吸附是多相催化过程,如水煤气变化和蒸气重整反应中重要的一步,因此其研究具有重要意义。由于包含9个自由度,以往的研究只能利用减维量子模型把体系的自由度限制在六个。今年初,该研究团队首次利用7 维量子动力学方法研究H2O在Cu(111)表面的解离吸附动力学 (Chemical Science, 2016, 7, 1840-1845),发现7维量子动力学结果和之前的6维结果相差很大,说明表面减维模型在描述此类反应时会带来较大的误差,因此非常有必要开展全维量子动力学研究。
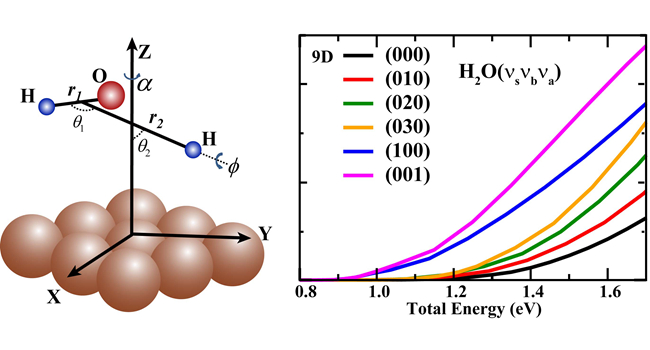
最近,该研究团队在其拟合的全维全域势能面上成功在全维(9维)水平计算了H2O在Cu(111)表面的解离吸附几率,从而首次实现了一个三原子分子在金属表面反应的全维量子动力学研究,被审稿人誉为“理解表面反应动力学的一个重要里程碑”。他们的研究发现全维量子解离几率与以往用减维模型得到的结果相差很大,证明只有全维量子计算才能精确描述此类反应。全维量子动力学还揭示了H2O的不同振动模式激发比平动能都能更有效地促进反应发生,并且效率明显要比减维模型得到的更为显著。该全维量子动力学研究也验证了该研究团队之前在双原子分子-表面散射中所发展的质心位点平均方法的准确性,表明该方法能广泛应用到多原子分子在金属表面的解离吸附动力学研究中,从而为精确模拟多原子分子-表面反应提供了一个重要的理论方法。
以上研究得到了国家自然科学基金委、科技部和中科院的支持。(文/图 傅碧娜)


